Getting Started¶
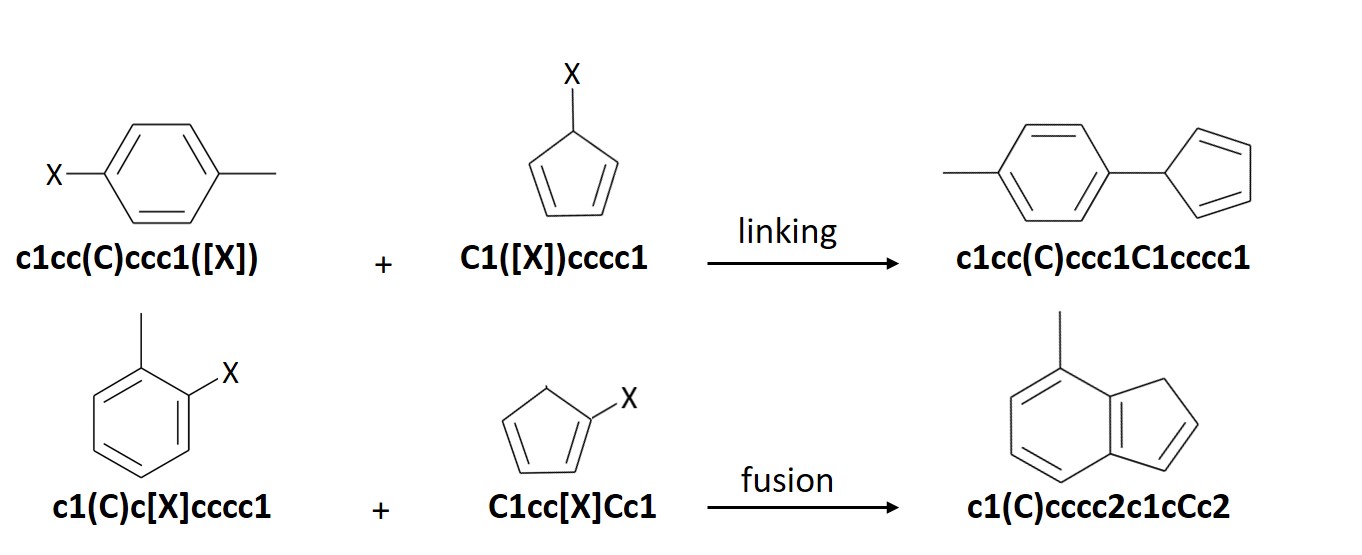
ChemLG generates molecules based on the following combinatorial ‘linking’ and ‘fusion’ schemes:
The ChemLG module is executed from the following command line:
chemlgshell -i ./config.dat -b ./building_blocks.dat -o ./output/
# for parallel runs:
srun -n $SLURM_NPROCS --mpi=pmi2 chemlgshell -i config.dat -b building_blocks.dat -o ./
The file names used above are the default values that the program looks for. The -o handle is used to specify the output directory where all the output files will be dumped. As seen from the command line above, the user should provide two input files:
a file with the configuration/rules for generating the library with the -i handle
a file with all the initial building blocks with the -b handle
Templates for both the input files are located at https://github.com/hachmannlab/chemlg/tree/master/chemlg/templates
It is highly recommended that the user substitutes the required values directly into the respective templates for the input files in order to avoid any conflicts later while executing the program. For more specifics on how to make the input files, look at:
Smarter library generation via Genetic Algorithm¶
ChemLG’s genetic algorithm module allows for an optimized growth of a library. That is, a library can be generated such that new members are added to the library only if they conform to a given range of target properties. For example, a library with each of the members having a density greater than 1000 kg/m3. The smarter library can also be made to optimize multiple properties at once using the multi-objective Genetic Algorithm module.
In this case, the user should provide:
genetic algorithm config file (template available at https://github.com/hachmannlab/chemlg/tree/master/chemlg/templates
path to the python file that contains the cost function defined by –> def cost_function(): <code> . The cost function always receives an openbabel object of a molecule. Cost function may return more than one value for optimization.
Optional input. Only required when running genetic algorithm in batch mode. Provide the path to the csv file containing the following column headers: individual, fitness, smiles. If running batch mode for the first time, provide the value: ‘empty’
For example:
Define the objective function:
# File name: example_cost_function.py
from openbabel import pybel
# This 'objective' function always receives an openbabel object of a molecule ('mol_ob' in the example). Also, the name of the objective function should ALWAYS BE cost_function.
def cost_function(mol_ob):
mol2 = pybel.readstring("smi", "c1ccccc1")
tanimoto = mol_ob.calcfp()|mol2.calcfp()
# tanimoto is the desired property which is returned by the function
return tanimoto
Execute ChemLG with Genetic Algorithm:
chemlgshell -i ./config.dat -b ./building_blocks.dat -o ./output/ --ga_config ./genetic_algorithm_config.dat --cost_function ./example_cost_function.py
## if running genetic algorithm in batch mode, also provide the fitnesses of the individuals with the --fitnesses_csv flag.
For more information on Genetic Algorithm parameters, refer to:
Feasibility Analysis¶
Even with the user-defined constraints that help eliminate undesired molecules, the final library can still end up with molecules that are not synthetically feasible. To address this issue of feasibility, we have incorporated a code that ranks these virtual molecules against a database of commercially available compounds (obtained from MolPort) based on the similarity of their fingerprints. With an upper or lower bound to the Tanimoto index provided by the user, the code processes the final library to remove all the molecules that fall short of the given criteria.
The following similarity metrics are included in ChemLG:
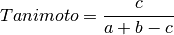
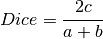
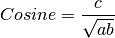
where, a is the number of features in structure 1,
b is the number of features in structure 2,
and c is the number of common features in structures 1 and 2.
Openpyxl is required as an additional dependency in ChemLG to run the feasibility analysis and can be installed as:
pip install openpyxl
The input to this function is the library generated by ChemLG and the cutoff Tanimoto index. This also gives the statistics of the generated library.
To run the feasibility analysis:
from chemlg.notebooks.feasibility import feasibility
The output.xlsx file contains a table of the molecules compared and their Tanimoto index.
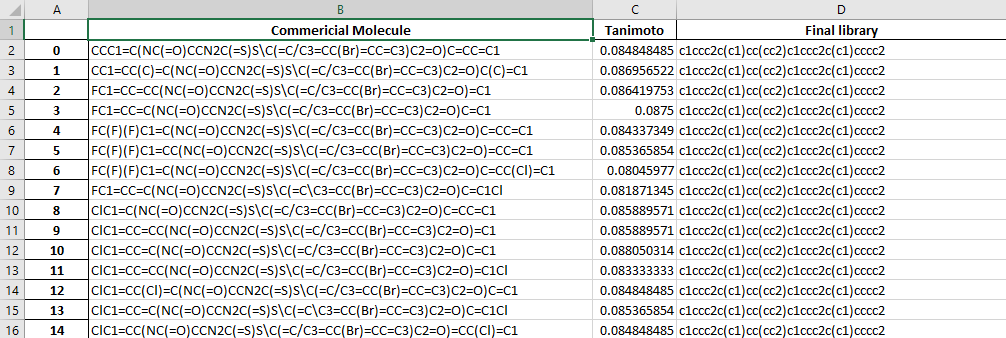
The dataframe.xlsx contains the table of molecule in the final library and the number of molecules which have a tanimoto index higher than the cut-off
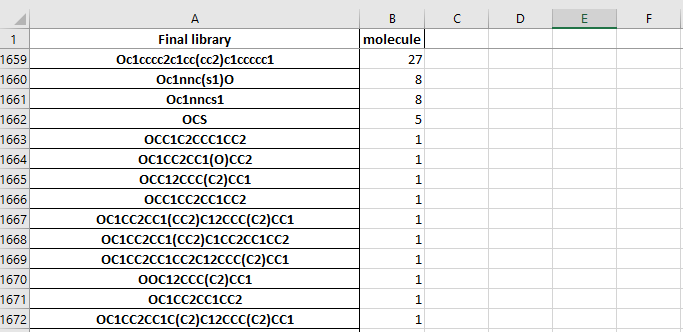
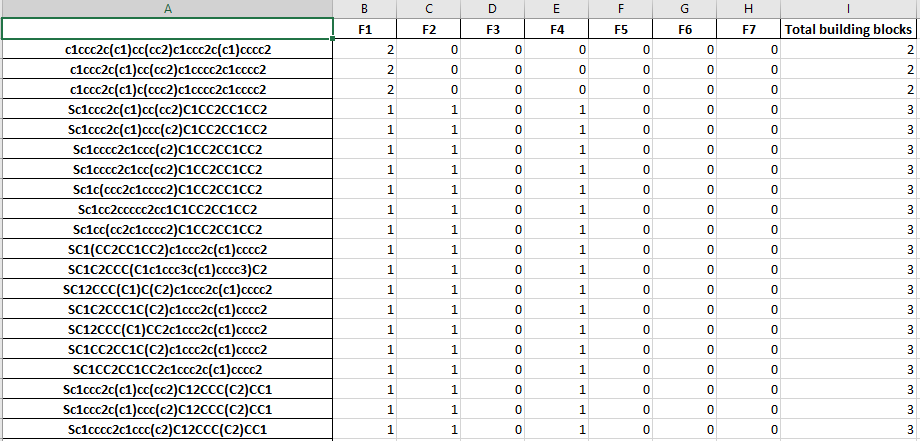
The stastictics.xlsx file cotains a table of the molecules and the number of each building block used in the molecule.